Esclerosis lateral amiotrófica (ALS)
¿Qué es la ALS?
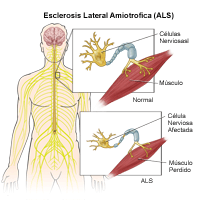 |
| Haga clic en la imagen para agrandarla |
La esclerosis lateral amiotrófica es un trastorno neurológico terminal caracterizado por una degeneración progresiva de las células nerviosas en la médula espinal y el cerebro. Con frecuencia se conoce como Enfermedad de Lou Gehrig (un famoso jugador de béisbol que murió de esta enfermedad), es uno de los trastornos más devastadores que afecta la función de los nervios y músculos.
La ALS no afecta el funcionamiento mental o los sentidos (como ver u oír), y no es contagiosa. Actualmente, no hay cura para la esclerosis lateral amiotrófica.
¿Cuán común es la ALS?
De acuerdo a la ALS Association (Asociación de la ALS), actualmente las siguientes estadísticas corresponden a la ALS:
-
La mayoría de personas que desarrollan ALS se encuentran en las edades de 40 y 70 años aunque la enfermedad puede ocurrir a una edad más temprana.
-
La edad promedio para el comienzo de la ALS es 55 años.
-
La ALS ocurre en todo el mundo sin respetar fronteras raciales, étnicas o socioeconómicas.
-
La ALS afecta a tantos como 30,000 estadounidenses con 5,000 casos nuevos diagnosticados en los EE.UU. cada año.
¿Cuáles son los tipos diferentes de ALS?
Hay tres clasificaciones conocidas de ALS, incluyendo las siguientes:
-
Esporádica. La forma más común de ALS en los EE.UU., que involucra del 90 al 95 por ciento de todos los casos. Estos casos ocurren aleatoriamente, sin ninguna causa conocida, y no existe asociación con personas en la familia con la enfermedad.
-
Familiar. Sugiere que la enfermedad es heredada y representa un número pequeño de casos en los Estados Unidos, cerca del 5 al 10 por ciento.
-
Guamano. Una incidencia extremadamente alta de ALS se observó en Guam y los Territorios en Fideicomiso de las Islas del Pacífico en los años 1950.
¿Cuáles son los síntomas de la ALS?
Los pacientes que sufren de ALS experimentan inicialmente debilidad en uno de sus miembros que se desarrolla en cosa de días o, más comúnmente, unas pocas semanas. Luego, de varias semanas a meses después, la debilidad se desarrolla en otro miembro. Algunas veces el problema inicial puede ser uno de habla incoherente o dificultad para tragar.
Sin embargo, a medida progresa la ALS, se advierten más y más síntomas. Los siguientes son los síntomas más comunes de la ALS. Sin embargo, cada persona puede experimentar los síntomas de manera diferente. Los síntomas pueden incluir:
-
Sacudidas y calambres de los músculos, sobre todo en las manos y los pies.
-
Pérdida de control de la motricidad en las manos y brazos.
-
Incapacidad en el uso de los brazos y piernas.
-
Tropezones y caídas
-
Dejar caer objetos
-
Fatiga permanente
-
Periodos incontrolables de risa o llanto
-
Habla pastosa o arrastrando las palabras y dificultad para proyectar la voz.
A medida progresa la enfermedad, los síntomas pueden incluir:
-
Dificultad para respirar
-
Dificultad para tragar
-
Parálisis
Los síntomas de la ALS pueden parecerse a otras condiciones o problemas médicos. Siempre consulte con su médico para un diagnóstico.
¿Cómo se diagnostica la ALS?
Además de un historial médico completo y examen físico, los procedimientos de diagnóstico para la ALS pueden incluir los siguientes:
-
Pruebas de laboratorio (incluyendo estudios de la sangre y la orina y pruebas del funcionamiento de la tiroides)
-
Biopsia de los músculos y/o nervios. Un procedimiento realizado para extirpar tejido o células del cuerpo para ser examinados bajo un microscopio.
-
Punción espinal (llamada también punción lumbar). Una aguja especial se coloca en la parte baja de la espalda, en el canal espinal; esta es el área alrededor de la médula espinal; luego se puede medir la presión en el canal espinal y el cerebro. Una pequeña cantidad de fluido espinal cerebral (CSF) se puede retirar y enviar para pruebas para determinar si hay una infección u otros problemas. El CSF es el fluido que baña el cerebro y la médula espinal.
-
Rayos X. Una prueba diagnóstica que usa rayos de energía electromagnética invisible para producir imágenes de los tejidos internos, huesos y órganos sobre una película.
-
Imágenes por resonancia magnética (MRI). Un procedimiento de diagnóstico que utiliza una combinación de imanes grandes, radiofrecuencias y una computadora para producir imágenes detalladas de órganos y estructuras dentro del cuerpo.
-
Pruebas electrodiagnósticas, como la electromiografía (EMG) y estudio de conducción nerviosa (NCS). Estudios que evalúan y diagnostican trastornos de los músculos y neuronas motrices. Se insertan electrodos en el músculo, o se colocan en la piel encima de un músculo o grupo de músculos, y se registra la actividad eléctrica y respuesta del músculo.
¿Cuál es el tratamiento para la ALS?
El tratamiento específico para la ALS será determinado por su proveedor de atención médica con base en:
-
Su edad, salud global e historial médico
-
Extensión de la enfermedad
-
Su tolerancia a medicamentos específicos, procedimientos o terapias
-
Las expectativas del tratamiento de la enfermedad
-
Su opinión o preferencia
Para la mayoría de personas con ALS, el tratamiento principal puede involucrar el manejo de síntomas, y puede incluir terapias físicas, ocupacionales, del habla, respiratorias y nutricionales. Algunos medicamentos y/o terapia de calor e hidromasajes pueden ayudar a aliviar los calambres musculares. El ejercicio, aunque se recomienda con moderación, puede ayudar a mantener la fortaleza y funcionamiento muscular.
No existe tratamiento comprobado para la ALS. Sin embargo, el Rilutek aprobado por la FDA, el primer fármaco que ha prolongado la supervivencia de personas con ALS.
El manejo de los síntomas de la ALS es un proceso que puede ser desafiante para personas con la condición, sus cuidadores y el equipo médico. Sin embargo, es importante saber que hay muchos recursos comunitarios disponibles para apoyo y asistencia.
Los investigadores están realizando estudios para aumentar su comprensión de los genes que pueden causar la enfermedad, mecanismos que pueden desencadenar la degeneración de las neuronas motrices en la ALS, y enfoques para detener el progreso que conduce a la muerte celular.